
Startseite | Mitarbeiter | Forschung | Lehre | Veranstaltungen | Anfahrt | Intranet
Deutsch | English
Startseite | Mitarbeiter | Forschung | Lehre | Veranstaltungen | Anfahrt | Intranet
Deutsch | English
Modern computational approaches based on density functional theory offer opportunities for exploring and understanding complex catalytic processes at a microscopic scale. Electronic structure calculations provide experimentalists with much needed insights into the chemical and physical processes that determine the properties of different types of catalysts.
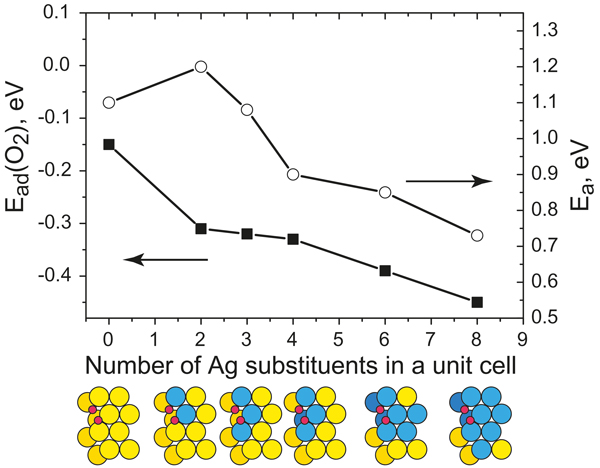 Adsorption energies, Ead, of O2 (squares) and the barrier height, Ea, for O2 dissociation (circles) as a function of the number of Ag impurity atoms in a unit cell. Transition state structures for O2 dissociation are schematically depicted.
Adsorption energies, Ead, of O2 (squares) and the barrier height, Ea, for O2 dissociation (circles) as a function of the number of Ag impurity atoms in a unit cell. Transition state structures for O2 dissociation are schematically depicted.
The project aims at understanding the mechanisms of oxidation reactions on gold-based catalysts such as nanoporous gold (npAu) at a microscopic level. It has been debated whether the catalytic properties of npAu are mainly due to structural features, i.e., the high number of low coordinated surface atoms. Experiments provided evidence that residual silver (present in very small concentrations) is crucial in terms of O2 activation.
We have studied the fundamental aspects of Au catalysis using theoretical models represented by the flat Au(111) and the kinked Au(321) slabs with and without Ag impurities. The theoretical investigation of co-adsorption and reaction between CO and atomic O on the kinked Au(321) surface allowed us, for example, to rationalize the formation of CO2 above 200K, observed in temperature programmed desorption studies of npAu. We have shown that Ag atoms incorporated into gold surfaces can facilitate the adsorption and dissociation of molecular oxygen on them. Therefore, the presence of the low-coordinated gold atoms is essential but apparently not sufficient for the catalytic activity of gold.
Lyudmila Moskaleva
References:
Cooperation partners: Thorsten Klüner, Oldenburg, Germany; Konstantin Neyman, Barcelona, Spain
Funding: DFG
For detailed information contact Lyudmila Moskaleva